
網膜色素変性症
網膜色素変性症は視細胞や網膜色素上皮の遺伝子の突然変異により生じる緩徐進行性遺伝性疾患である。網膜色素変性症は杆体ジストロフィと錐体杆体ジストロフィを指し、視細胞のうち杆体細胞のみの変性を杆体ジストロフィ、杆体細胞と錐体細胞両者の変性を杆体錐体ジストロフィと称する。
日本では3000~8000人に1人の割合で発症し、成人視覚障害原因の第3位として知られている。典型例は10~20歳代に夜盲で発症し、徐々に周辺視野が障害され、最終的には中心視力低下から完全に失明に至る。
しかし、80歳になっても中心視力良好な例も存在するなど、その遺伝様式等によって異なるとされている。小児期は視力低下よりも夜盲や視力低下を主訴に来院することが多い。
網膜色素変性症の遺伝様式
孤発例:56%
常染色体優性遺伝:17〜40%
常染色体劣性遺伝:25〜60%
X連鎖性遺伝:2〜20%
常染色体優性遺伝では、他の遺伝様式に比べて発症年齢が遅く、良好な視力を保つ可能性が高い。現在、類縁疾患を含めると80種以上の原因遺伝子が見つかっている。
日本で最も頻度が多いの原因遺伝子はEYS、次いでUSH2Aである。また、原因遺伝子として優性遺伝ではRHO遺伝子、劣性遺伝ではUSH24遺伝子、X連鎖劣性遺伝ではRPGR遺伝子が原因遺伝子として多いとされる。
そして、網膜色素変性症は難聴など全身疾患を伴うものも含まれる現在までに60以上の遺伝子が知られている。全身症状を伴うものとして30以上の症候群があるが代表的なものを示す。
1.Usher症候群
感音難聴を伴い、常染色体潜性遺伝の網膜色素変性症の20~40%を占めるという報告がある。
type1
先天性の重度難聴があり、10歳前後に発症する。両側の前提機能障害を伴う。原因遺伝子として、CDH23、MYO7A、PCDH15、USH1C、CIB2、USH1Gが報告されている。
type2
先天性の高音障害型の難聴があり、思春期以降に発症する。前庭機能は正常である。原因遺伝子としてUSH2A、GPR8、DFNB31が報告されている。
type3
進行性の難聴を伴う。RPは発症しても他のタイプよりは軽度である。原因遺伝子としてCLRN1とPDZD7が報告されている。
2.Bardet-Biedl症候群
肥満、精神発達遅滞、多指症、性腺機能低下症、腎奇形(慢性腎障害)を伴う。常染色体潜性遺伝である。海外のコホート研究では網膜色素変性症の5~6%を占めるという報告がある。
3.Kearns-Sayre症候群
眼筋麻痺、網膜変性、心筋症、短躯、筋力低下、難聴、糖尿病など様々な症状を合併しうる。
4.Leber congenital amaurosis
生下時から眼振、低視力を認める網膜ジストロフィ全てを含む疾患群のことを指す。
5.Alström症候群
Alström症候群は若年発症のRPもしくはLeber先天盲、白内障、肝機能障害、高脂血症、低身長、性腺機能障害、呼吸器障害、腎機能障害、扁平足、インスリン抵抗性2型糖尿病を合併する常染色体潜性遺伝の疾患である。ALMS1遺伝子の両アレル性変異によって発症する。ALMS1タンパクは、視細胞内小器官である繊毛に発現し、視細胞内節から外節へのタンパク輸送に重要である。
6.Senior-Loken症候群
Senior-Loken症候群は若年発症のRPもしくはLeber先天盲に、ネフロン癆(腎髄質に嚢胞形成を認め、徐々に末期腎不全になる)を合併する常染色体潜性遺伝の疾患である。原因遺伝子としてNPHP1、INVS、NPHP3などが報告されている。
その他の症候性の遺伝性網膜ジストロフィ
脳回状脈絡網膜萎縮:常染色体潜性遺伝の先天性代謝異常症で、ビタミンB6依存性オルニチンアミノ基転移酵素活性の低下による高オルニチン血症、高オルニチン尿症を特徴とする。OAT遺伝子の両アレル性変異によって発症する。幼少期の夜盲があり、進行性の脈絡網膜編成をきたす。治療は低アルギニン食、低蛋白食の食事によって、オルニチン値を低下させる。また、ビタミンB6の投与でも低下することがある。
筋強直性ジストロフィⅠ型:常染色体顕性遺伝で、筋強直現象や筋力低下、前頭部脱毛、白内障、遺伝性網膜疾患、難聴を特徴とする。トリプレットリピート病で、DMPK遺伝子3’YTRのCTGリピートの異常伸長によって引き起こされる。
ミトコンドリア病:母系遺伝で、黄斑ジストロフィを合併することがある。
脊髄小脳変性症:全体の2/3が孤発例、1/3が遺伝性で、常染色体顕性遺伝が最も多い。特に、Machado-Joseph病、SCA6、SCA31、歯状核赤核・淡蒼球ルイ体萎縮症(DRPLA)の頻度が高い。SCA1で黄斑ジストロフィ、SCA7で錐体ジストロフィを合併した例がある。
網膜色素変性症の症状
通常は杆体細胞(網膜周辺に多く存在し、明暗を認識する視細胞) の変性により夜盲を生じる。徐々に杆体細胞の変性が進行すると、輪状暗点(周辺部視野障害)から始まり、求心性視野障害をきたすようになる。ただし、視野障害は緩徐に進むとされ、発症から失明に至るまでには数十年を要する。
網膜色素変性症に伴う合併症
- 白内障:約半数で見られる。若年時から中央の後嚢下混濁を認めることが多い
- 閉塞隅角緑内障、星状硝子体症
- 黄斑浮腫、黄斑前膜(数%)
網膜色素変性症の診断
A. 自覚症状
夜盲、求心性視野障害、視力低下(※)
※幼少期~成人までに夜盲、次第に中間周辺部の視野欠損⇒周辺部視野が消失し、中心視野のみが残存するようになる。
B. 他覚症状
1.眼底所見
網膜血管狭細、網膜色素上皮の変性萎縮、骨小体様色素沈着
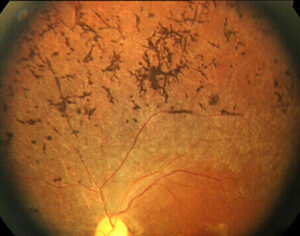
骨小体様色素沈着(Retina Macula Institute HPより引用)
2.光干渉断層計(OCT)
網膜外層の障害(周辺部から中心へ錐体視細胞外節端、網膜視細胞内節外節接合部(EZ line)、外境界膜の順)があるため、外顆粒層の菲薄化やEZ/IZ lineの消失を認める。また、黄斑浮腫(ME)、黄斑前膜を高率に認める。
※中心窩のIS/OSラインの描出が悪くなると、視力低下を認める傾向がある。
3.フルオレセイン蛍光眼底造影検査(FA)
造影初期:脈絡膜大血管が透見可能で、一致して点状過蛍光がみられる(window defect)
4.網膜電図(ERG)
確定診断にはERGを行う。初期から消失型もしくはa波、b波の振幅低下を示す。ただし、杆体機能が低下し、その後錐体機能が低下するため、早期の症例では錐体系応答(錐体応答あるいはflicker応答)はわずかに反応が検出できることが多い。

5.視野検査
典型的には求心性視野障害を示す。初期は島状の暗点となり、進行するとそれらがつながって輪状暗点となる。
6.眼底自発蛍光(FAF)
変性が進行し、色素上皮が消失した部位は低蛍光に写る。EZラインの存在する部位と消失している部位の境界は過蛍光に写る。低蛍光部位はGoldmann視野の狭窄、暗点と一致しやすい。j
上記を踏まえ、難病情報センターに提示されている診断基準を示す。

また、重症度も下記のようになされる。

網膜色素変性症の治療方法
RPE関連網膜症に対する遺伝子治療を除いて、現在有効な治療方法はないとされているが、病期に応じて様々な治療戦略が考えられている。
1.サプリメント
- ビタミンA:進行を抑制する場合もあれば、原因遺伝子によっては進行を助長する恐れがある。ビタミンA15000IU/日投与で、5年後にERGの錐体反応の減少を有意に抑制できるという報告がある。
- DHA、ルテイン:黄斑部視細胞の酸化ストレスから保護するため進行抑制に働くとされる。
- N-アセチルシステイン(NAC):NACはアセトアミノフェン過剰摂取時の解毒薬として国内で薬事承認されている。NAC1日3600㎎内服は現在第Ⅲ相臨床試験が実施されている。
※VitA+ルテイン12㎎/日で、中間周辺部視野進行抑制という報告がもある。
2.遺伝子治療
RPE関連網膜症に対する遺伝子治療が保険収載された。また、RPGR遺伝子異常による網膜色素変性症に対してはウイルスベクターを用いた遺伝子治療が有効な可能性がある。
3.神経保護
ウノプロストン点眼、Ca拮抗薬のニルバジピン、毛様神経栄養因子あるいは色素上皮由来因子などの視細胞保護効果のある物質を用いた臨床試験が実施されている。
4.人工網膜
欧米では臨床応用されている。外部に設置されたカメラと網膜をつなぎ刺激を送る。
5.網膜再生移植術
iPS細胞由来の視細胞移植で改善する可能性がある。
6.その他合併症の治療
白内障などを合併することがあるため、白内障が強ければ白内障手術を行う。このように合併症の治療も併せて行っていく。
網膜色素変性症の鑑別疾患
- コロイデレミア
- 脳回状脈絡網膜萎縮
- 癌関連網膜症
- 梅毒
- ぶどう膜炎
- アルコール
- 薬物依存症
参考文献
- 今日の眼疾患治療指針第3版
- 眼科学第2版
- 日本眼科学会HP
- 難病情報センターHP
- 日本眼科学会専門医制度生涯教育講座総説64『小児の網膜疾患と網膜電図』
- 黄斑疾患診療AtoZ
- あたらしい眼科 Vol.42, No.3, 2025
- Visual Outcomes in Japanese Patients with Retinitis Pigmentosa and Usher Syndrome Caused by USH2A Mutations
- Genetic characterization of 1210 Japanese pedigrees with inherited retinal diseases by whole-exome sequencing
- Progress of macular atrophy during 30 months’ follow-up in a patient with spinocerebellar ataxia type1 (SCA1)
- Somatic instability of expanded CAG repeats of ATXN7 in Japanese patients with spinocerebellar ataxia type 7
- Retina Medicine vol.14 no.1 2025
関連記事






